- What is the CGI?
- How is the CGI framework?
- Which type of alterations does the the CGI interprets?
- How are the driver events identified?
- What is OncodriveMUT?
- What is the performance of OncodriveMUT in the identification of driver mutations?
- How are actionable events identified?
- What is the cancer bioMarkers database?
- Can CGI be used to interpret more than one tumor sample at a time?
- Why is it necessary to select a cancer type when interpreting a sample(s)?
- Why is there a cancer taxonomy?
- How are the mutations mapped?
- How is the CGI output provided?
- Which is the CGI license?
- How can I input alteration data?
- Which mutations formats are accepted?
- How long does it take to have my CGI analysis finished?
- Are there any platform requirements for the mutation data?
- What happens if a mutation can not be mapped to the reference genome?
- Do I need to login to run a CGI analysis?
- Can I download CGI results as a file?
- Are my CGI results private?
- Can I share my CGI results?
- Can I provide feedback on CGI?
- Is there a REST API?
What is the CGI?
Cancer Genome Interpreter (CGI) is designed to support the identification of tumor alterations that drive the disease and detect those that may be therapeutically actionable. CGI relies on existing knowledge collected from several resources and on computational methods that annotate the alterations in a tumor according to distinct levels of evidence.
How is the CGI framework?
With a list of genomic alterations in a tumor of a given cancer type as input, the CGI automatically recognizes the format, remaps the variants as needed and standardizes the annotation for downstream compatibility. Next, it identifies known driver alterations and annotates and classifies the remaining variants of unknown significance. Finally, alterations that are biomarkers of drug effect are identified according to current evidences.
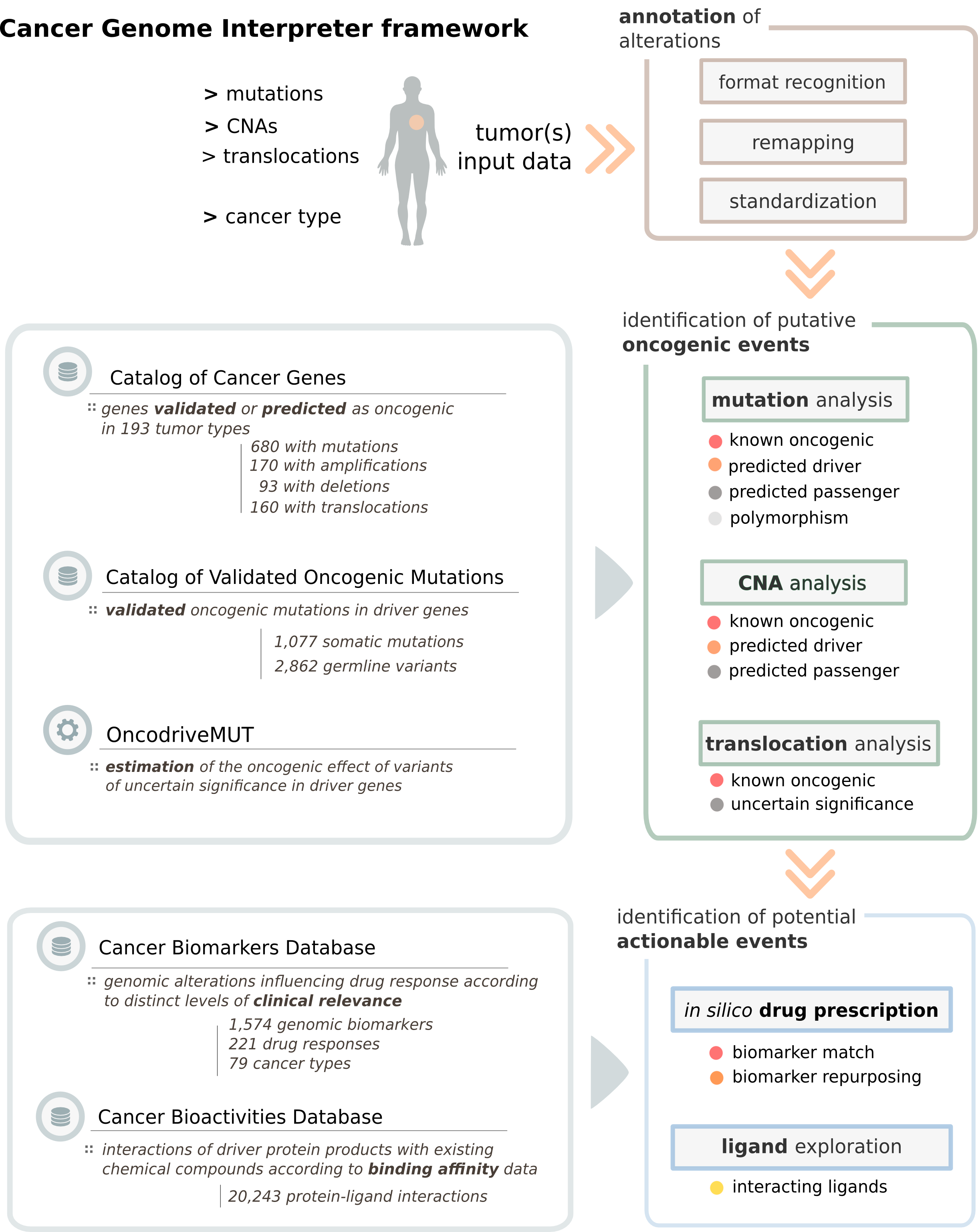
Schema summarizing the CGI framework
Which type of alterations does the the CGI interprets?
CGI analyses mutations (single nucleotide changes and small insertions/deletions), copy number alterations (gene amplifications and deletions) and translocations.
How are the driver events identified?
Alterations that are clinically or experimentally validated to drive tumor phenotypes –previously culled from public sources-- are identified by the CGI, whereas the effect of the remaining alterations of uncertain significance are predicted using in silico approaches, such as OncodriveMUT (for mutations).
What is OncodriveMUT?
OncodriveMUT is a bioinformatics method to identify the most likely driver mutations of a tumor. Its main innovation with respect to other existing tools with a similar purpose is the incorporation of features characterizing the genes (or regions within genes) where the mutations occur, derived from the analysis of cohorts of tumors (6,792 samples across 28 cancer types) and samples from healthy donors (60,706 unrelated individuals). This knowledge is combined with features that describe the impact of the mutation on the function of the protein it affects via a set of heuristic rules to predict the effect of the mutations of uncertain significance.
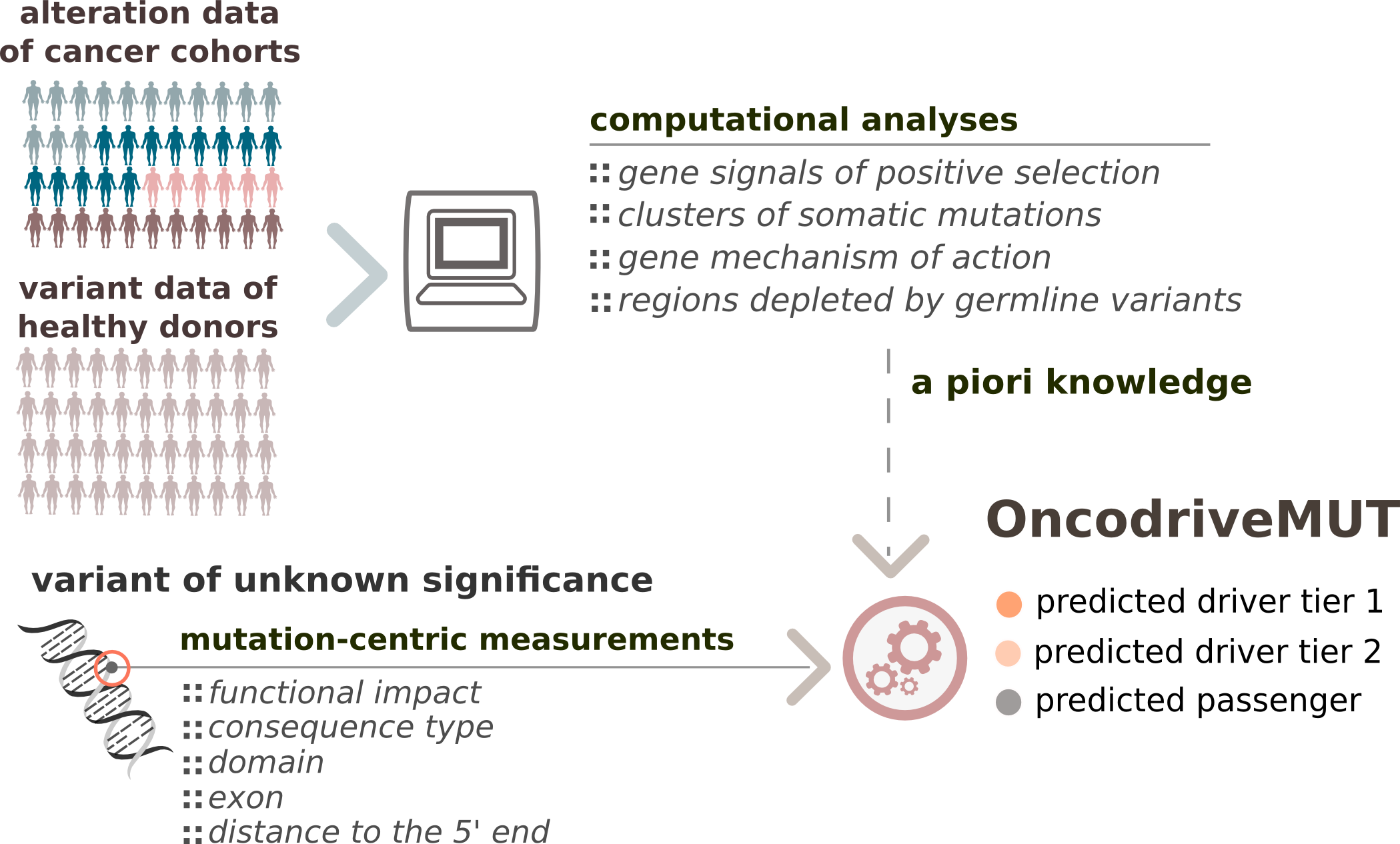
Schema summarizing the OncodriveMUT method framework
What is the performance of OncodriveMUT in the identification of driver mutations?
To benchmark the performance of OncodriveMUT, we compiled a set of known driver somatic mutations (n=1,077) and cancer-predisposing germline variants (n=2,819) (positive set) and another set of validated non-pathogenic somatic mutations (n=241) and common (major allele frequency > 1%) polymorphisms (n=1,006) affecting cancer genes (negative set). Noteworthy, only protein-affecting mutations in cancer genes have been included, a ground in which the distinction between driver and passenger mutations is more challenging than across all human genes. We found that OncodriveMUT distinguishes between the positive and negative sets of variants in cancer genes with an accuracy of 0.91 and Matthews correlation coefficient of 0.78.
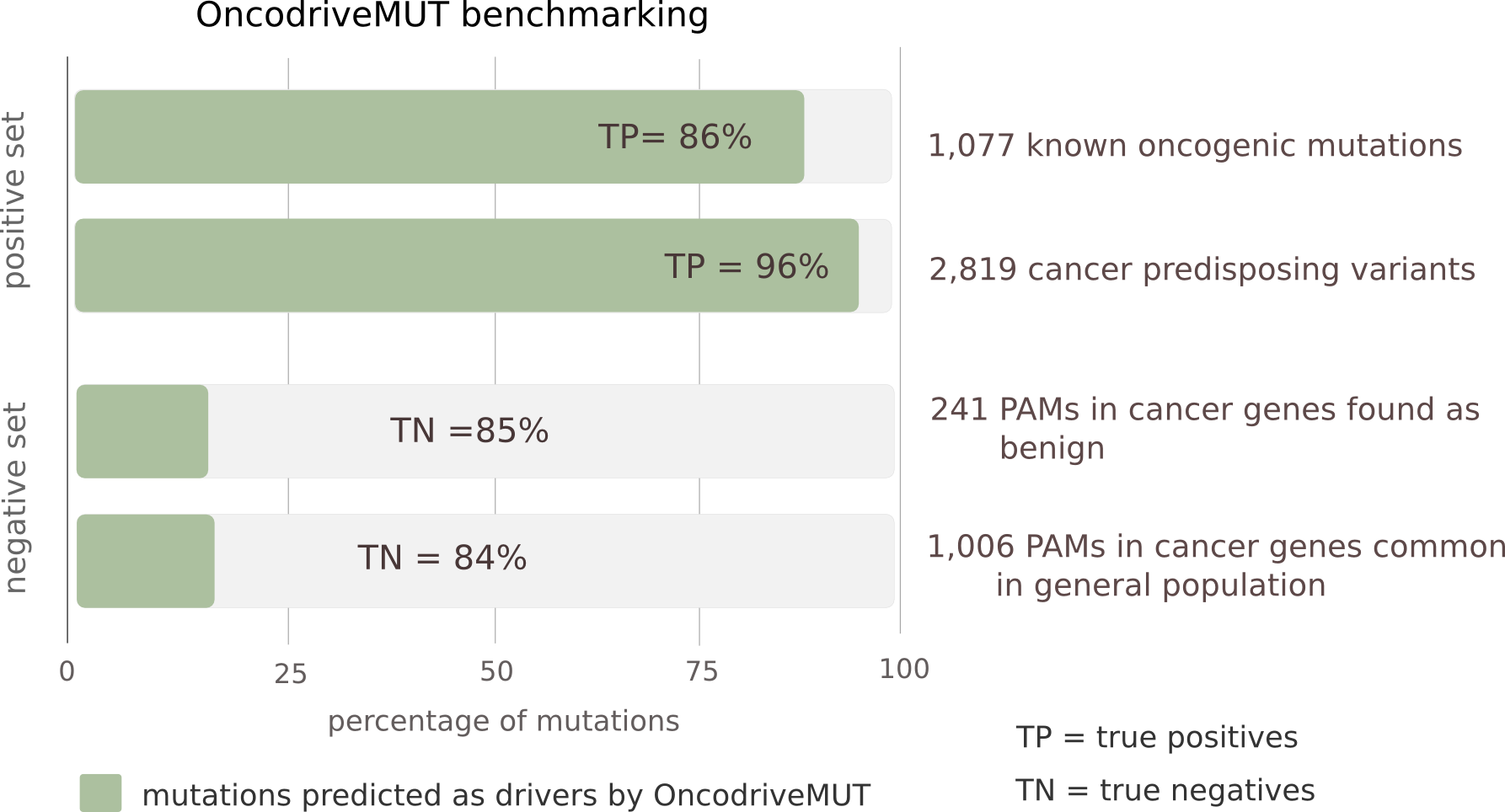
Performance of OncodriveMUT to distinguish bona fide driver and passenger protein-affecting mutations (PAM) among cancer genes
How are actionable events identified?
To assess the relevance of the alterations as biomarkers of drug response, the CGI relies on an in-house database (cancer bioMarkers-db) of genomic events that influence the response of the tumor to a drug (sensitivity, resistance or severe toxicity), with varying levels of clinical relevance according to state-of-the-art knowledge, ranging between standard-of-care guidelines and the evidences derived from preclinical assays. Chemical interactions of other compounds that bind altered genes in the tumor are also mined and retrieved by the CGI (cancer bioActivity-db).
What is the cancer bioMarkers database?
The cancer biomarkers db integrates manually collected genomic biomarkers of drug sensitivity, resistance and severe toxicity. These biomarkers are classified by the cancer type in which they have been described according to different levels of clinical evidence supporting the association. The database is available for access and feedback by the community at www.cancergenomeinterpreter.org/biomarkers. The aggregation, curation and interpretation of the biomarkers follow the standard operating procedures developed under the umbrella of the H2020 MedBioinformatics project, thus ensuring the mid-term maintenance of these resources. The feedback from the community is also facilitated through the CGI web interface. Nevertheless, access to this type of data is both crucial for the advance of cancer precision medicine and highly complex to be comprehensively covered and updated by a single institution. This is why the Variant Interpretation for Cancer Consortium, under the Global Alliance for Genomics & Health framework (Global Alliance for Genomics and Health et al. 2016), has recently been launched with the aim to unify the curation efforts in several institutes, including ours.
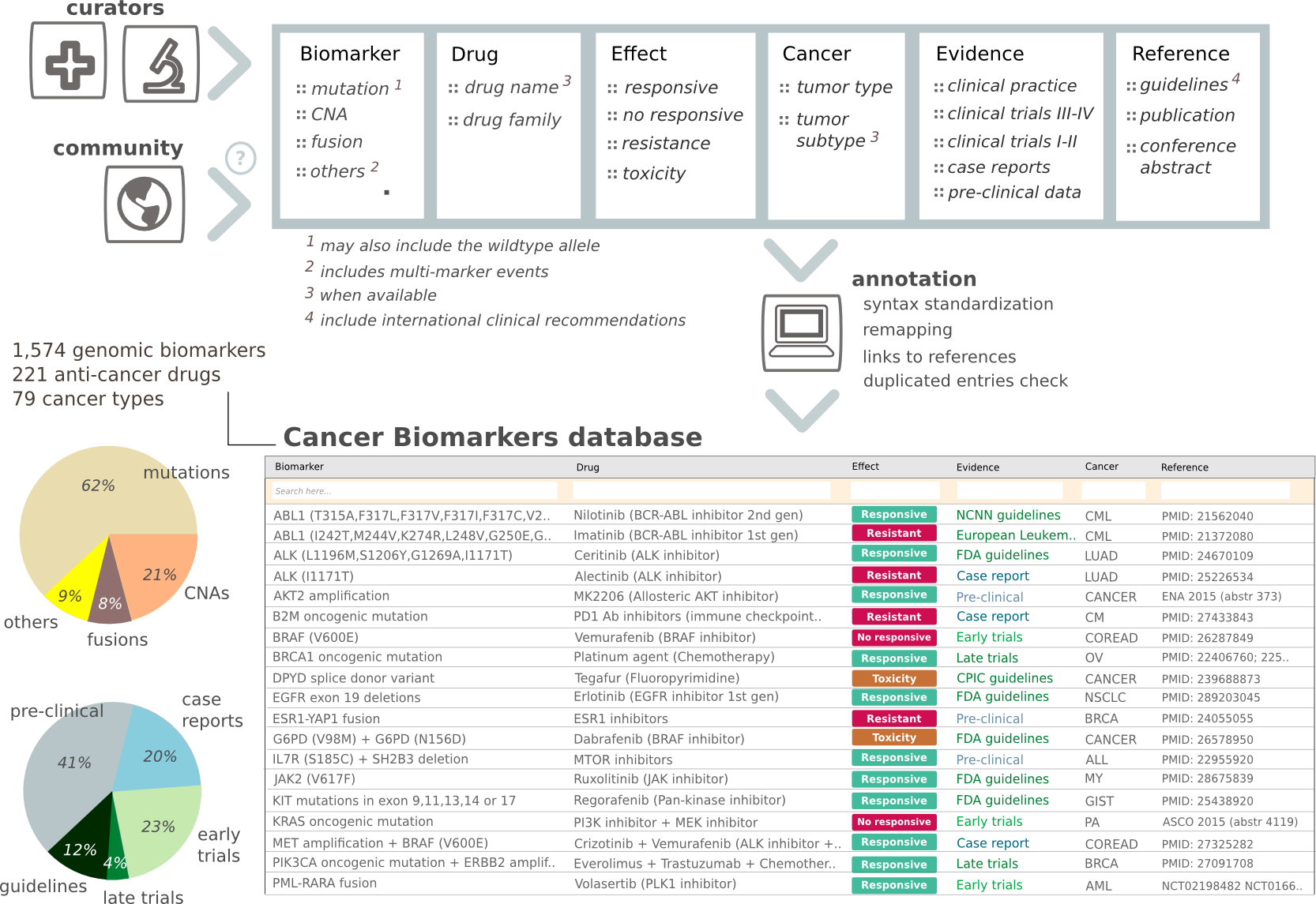
Schema of the cancer biomarkers database
Can CGI be used to interpret more than one tumor sample at a time?
A CGI execution can include the alterations detected in one or more tumor samples, as far as they correspond to the same cancer type. To distinguish the results of several samples you need to provide one column in the mutation file with a sample identifier.
Why is it necessary to select a cancer type when interpreting a sample(s)?
CGI prediction of driver mutations via OncodriveMUT employs knowledge on the role of each gene and gene region in specific cancer types, obtained from the analysis of large sequenced cohorts of tumors. Of note, when this knowledge is not available for the cancer type under analysis, CGI runs a generic interpretation using pan-cancer information on the genes. In addition, the identification of known driver alterations as well as the biomarkers of drug response take into account the match between the tumor type in which they have been described and the one of the sample under analysis.
Why is there a cancer taxonomy?
The taxonomy is used to integrate the information on subtypes of the tumor selected by the user when appropriate. For instance, if the user selects non-small cell carcinoma as the tumor type of the sample(s) to interpret, the a priori knowledge specific to lung squamous carcinoma and lung adenocarcinoma will be used. We're currently working in harmonizing this taxonomy with international efforts as disease-ontology.org
How are the mutations mapped?
Both the mapping from DNA to protein change and from protein to DNA change are performed by TransVar. Of note, the system currently uses Ensembl version 70, although this will be updated in next CGI releases.
How is the CGI output provided?
When the CGI is run via web, the results of the are provided via interactive reports. These web reports can be interactively browsed and configured by the user. A mutation analysis report contains the annotations of all variants, which empowers the user's review, and the labels for those known or predicted to be drivers by OncodriveMUT. A biomarkers-match report contains the putative biomarkers of drug response found in the tumor organized according to distinct levels of clinical relevance.

CGI output example
Which is the CGI license?
The results generated by the CGI output are under a Creative Commons Attribution-NonCommercial 4.0 (BY-NC) license. When referring to the datasets, any result generated by the CGI framework or the CGI resource itself, please cite: Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations; doi: https://doi.org/10.1101/140475 and the main page https://www.cancergenomeinterpreter.org.
How can I input alteration data?
The user can type the alterations (mutations, copy number alterations and translocations; see format) within the alteration box (one alteration per line) or upload them in one or more alteration files (all these files will be pooled together to run a single analysis).
Which mutations formats are accepted?
Check our format help page.
How long does it take to have my CGI analysis finished?
The execution time depends on i) how long the job takes to get a slot in the cluster, ii) the time required by the data structures used by the CGI to be loaded, and iii) the number of entries to be analysed. Therefore, to get the results may take a while even if only few mutations are submitted. If you want to have an overview of the CGI results, you can check out the examples
Are there any platform requirements for the mutation data?
CGI interprets mutations obtained using any sequencing platform (e.g. exome sequencing, whole-genome sequencing or a gene panel) or calling pipeline. Of note, if the data include germline variants (i.e. the mutations have been not called only for somatic events), this can add some noise to the CGI driver analysis, although the oncogenic events (both known and predicted) are expected to be highly enriched for true somatic mutations. Also, germline variants that are known as cancer related (e.g. BRCA variants that predispose to breast tumors) are expected to be detected by the CGI. Of note, common polymorphisms (AF>1%) identifications are included in the CGI.
What happens if a mutation can not be mapped to the reference genome?
Those mutations that cannot be mapped (if any) will be filtered out from the CGI analysis. Reasons of incorrect mapping include an unrecognizable format or incorrect mutation information (e.g. an invalid reference allele or chromosome position). If the number of unmapped mutations exceeds 30% of all mutations submitted, the system assumes that the data is not reliable and does not proceed with the interpretation. We also remove those mutations mapping in non-exonic regions. Of note, unmapped mutations are accessible as a separate file for further review.
Do I need to login to run a CGI analysis?
No, the CGI can be used without login. The results of the run will be available for 24 hours at the link provided by the system upon submission. On the other hand, if the user logs into the system and click the button 'Save' in the analysis box, it won't be removed after the 24h. Note that the login process only requires a valid email address and access is immediately granted. Any analysis launched with a logged user are automatically saved.
Can I download CGI results as a file?
Yes, once the interpretation is completed, the results can be downloaded as tab-separated files through the corresponding button.
Are my CGI results private?
To prevent unauthorized access or disclosure, to maintain data accuracy, and to ensure the appropriate use of information, CGI uses a range of reasonable physical, technical, and administrative measures to safeguard your Personal Information, in accordance with current technological and industry standards. In particular, all connections to and from our website are encrypted using Secure Socket Layer (SSL) technology. CGI never has access to your password and uses a trusted third party (gmail, mozilla persona, yahoo...) protocol to authenticate you. While the analyses are running and accessible from our web server they are stored in our private servers. When you remove an analysis it will completely and permanently removed from our servers, and we do not keep any copy. We do not share our files with third parties.
Can I share my CGI results?
Yes. Once the analysis is completed, the user may share the results by clicking the 'Share' button. But be aware than then anyone with the URL will be able to access your analysis. We plan to add support to be able to give access only to certain users.
Can I provide feedback on CGI?
For any comment, suggestion or error report, please contact with bbglab@irbbarcelona.org
Is there a REST API?
Yes, find all the details in this link: www.cancergenomeinterpreter.org/rest_api